



In-Depth
Emergency use authorization (EUA) of medical products during the COVID-19 pandemic including a look ahead at possible EUA of a COVID-19 vaccine
Recent media reports, government statements and expert opinions have left many in the U.S. expecting a possible emergency use authorization (EUA) from the U.S. Food and Drug Administration (FDA) for a COVID-19 vaccine in the coming months. This brief will serve to provide an overview of the research and regulatory steps necessary for a vaccine or other unapproved medical product to be given any type of emergency use authorization. It will also look at the current landscape of candidate vaccines in phase 3 trials in the U.S..
Mechanisms to allow for emergency use of unapproved medical products (drugs including antivirals and antidotes, biologics including vaccines and blood products, and devices including diagnostic tests and personal protective equipment) or for emergency use of approved medical products for conditions outside their approval were established in 2004 through amendments to the Federal Food, Drug and Cosmetic Act of 1937. Since 2004, additional legislation has solidified the EUA as part of the FDA’s role in reviewing the safety and efficacy of medical products and strengthening the nation’s public health preparedness and response. Though there is no precedent for an EUA being issued for a new unlicensed vaccine, an EUA was issued in 2005 to allow an approved anthrax vaccine licensed in 1970 to be used specifically for inhalation anthrax in people designated as high-risk for exposure. Several EUAs were issued in 2009 for public health and clinical activities around the H1N1 influenza response.
The EUA process can be used during times of national emergency in response to a chemical, biological, radiological or nuclear (CBRN) threat. Once a state of emergency is declared, certain regulatory requirements may be temporarily waived or modified to facilitate preparedness and response activities. During a public health state of emergency, as was declared due to the COVID-19 pandemic, public and private entities may request an EUA for medical products that are needed in an emergency to “diagnose, treat or prevent serious or life-threatening diseases or conditions caused by CBRN threat agents where there are no adequate, approved and available alternatives.”
So far during the COVID-19 pandemic, the FDA has issued dozens of EUAs. As of September 11, most (246) have been for diagnostic and serologic tests and assays. Twenty EUAs have been issued for personal protective equipment (PPE) and systems that play a role in infection prevention and control, and 26 EUAs have been issued for medical devices, such as ventilators and heart monitors. Only six drug or biologic products have been extended this emergency approval, three of which play a role in directly treating COVID-19. These are remdesivir—an antiviral, hydroxychloroquine sulfate (and chloroquine phosphate)—antimalarials initially thought to possibly improve outcomes from COVID-19, and convalescent plasma—a blood product obtained from recovered COVID-19 patients containing disease-fighting antibodies. The EUA for hydroxychloroquine sulfate (and chloroquine phosphate) has since been revoked due to a determination by the FDA that the drugs are “unlikely to be effective in treating COVID-19” and “in light of ongoing serious cardiac adverse events and other serious side effects.” The EUA for convalescent plasma has been met with much controversy due to a lack of data for its efficacy, and the FDA commissioner has since apologized for misstating the therapy’s potential to reduce the risk of death.
With no cure and limited therapeutic options, developing a vaccine that would prevent illness has been the center of a worldwide strategy to address the ongoing pandemic. There are currently at least 128 (other sources cite nearly 200) candidate vaccines under study, 36 of which are in clinical trials in humans. Of these, nine have advanced through dosage and safety testing to phase-3 trials where candidate vaccines are tested in large-scale studies recruiting tens of thousands of volunteers who receive either the candidate vaccine or a placebo or an unrelated vaccine to evaluate efficacy. Efficacy is determined once a certain number of people in the study develop disease, and an analysis is done to compare the incidence or attack rate in vaccinated versus unvaccinated people. Efficacy refers to how well the vaccine works in the ideal research conditions of such a trial.
For traditional vaccine approval, developers would typically wait for these phase-3 studies to reach their desired endpoints and be completed and submit their results to regulatory bodies for consideration of licensure. The traditional approval process once an application has been filed can take over a year.
For emergency use authorization, this process would need to be accelerated, and could be condensed into months. In late August, the FDA commissioner announced that a vaccine could be considered for EUA even before completing a phase-3 trial, based on preliminary data on safety and efficacy. The agency also issued guidance in June for industry to consider in their approach to developing and seeking licensure of a vaccine to prevent COVID-19. In this document, the FDA proposes minimum efficacy parameters that it would consider for a vaccine (50%, with 95% confidence that the true efficacy is at least 30%).
There is added pressure to get a vaccine EUA right. Although treatments used under emergency authorization are being given to seriously ill patients without other options for therapy, vaccines are designed to be given to healthy people in order to prevent disease. The balance of risks and benefits for a new vaccine could tip dramatically if a new product does not work well, and if it causes any harm.
It can take years for the safety profile of a new vaccine to be fully understood. Although the U.S. provides its residents with the safest most effective vaccines, this is in part due to the lengthy and rigorous safety and efficacy trials that are taken into consideration when licensing a new vaccine. Despite these efforts, very rarely, evidence emerges to reinforce the need for even more rigor. To date, only one vaccine has been removed from the U.S. market after licensure: Rotashield—a rotavirus vaccine which had been studied extensively prior to being approved and licensed. In fact, the vaccine had been studied for 28 years prior to being presented to regulatory bodies for licensure. It was not until the vaccine was used in large numbers in the general pediatric population that information about a potentially fatal complication became more apparent. The “climate” in which the vaccine’s safety was questioned was a turbulent one for vaccines overall, and this may have contributed to its removal from the market.
Operation Warp Speed is a public-private partnership founded in May 2020 to allow for the accelerated development, manufacturing and distribution of a COVID-19 vaccine. Alhough the initial concept aimed to have 300 million doses of vaccine delivered by January 2021, an updated goal states that the Operation aims to have the initial doses of vaccine available by January 2021. Authorities have consistently messaged that while a timeline will be set to accelerate manufacturing and distribution, the duration of phase 3 trials will remain unaffected. Accomplishing this will require that multiple phases of trial be run in parallel rather than in sequence in order to produce an overall shorter timeline while keeping standard durations expected for safety and efficacy studies.
Of the candidate vaccines involved in the partnership, a vaccine developed at the University of Oxford and licensed to the British pharmaceutical company AstraZeneca for further testing had received much attention as potentially leading the pack toward consideration for an EUA. This vaccine’s late stage trial was briefly placed on pause as a result of a serious suspected adverse event that required a safety review. This type of process is exactly what clinical trials are designed in-part to allow for. The Asta-Zeneca vaccine uses a common cold virus affecting chimpanzees to deliver proteins from the virus that causes COVID-19 in order to induce immunity in humans. In May, the AstraZeneca vaccine had already been in phase 3 trials in other parts of the world including Brazil, the U.K. and South Africa, when the U.S. Health and Human Services announced up to 1.2 billion dollars to support availability of 300 million doses in the U.S. In late August, the company announced plans to expand its phase-3, double-blind, placebo controlled trial to 30,000 people in the U.S. The recent adverse event happened in the U.K. and the trial there has since been restarted. On the National Library of Medicine’s clinical trial registry site, www.clinicaltrials.gov, the study protocol states that it anticipated primary completion data from its U.S. arm as soon as early December 2020 and completion in October 2022.
Other candidate vaccines which may be among the first to present safety and efficacy data for EUA consideration are Moderna’s mRNA vaccine co-developed by the National Institute of Allergy and Infectious Diseases and Pfizer/BioNTech’s mRNA vaccine, both of which began phase 3 studies in July 2020. Moderna’s recruitment was slowed to allow for improved recruitment of diverse segments of the U.S. population. Pfizer/BionTech have also announced that they may increase overall recruitment and increase diversity in study participants. It is still possible that data from international arms, as well as U.S. arms of these studies, could be presented to a regulatory vaccine committee which is scheduled to meet in mid-October 2020. This committee would give a recommendation for or against an EUA after reviewing the available evidence for safety and efficacy. If given emergency use authorization based on this early data, the developer would have about two months to meet the production and distribution timeline set by Operation Warp Speed.
In a recent discussion hosted by the Duke University Margolis Center for Health Policy, Peter Marks, the director at the FDA’s Center for Biologics Evaluation and Research, stated that any EUA for a vaccine would be treated more like an “EUA plus” which he described as a process falling between the requirements to meet criteria for a biologic EUA and the requirements of a traditional Biologic License Application (BLA). A BLA would be submitted and reviewed for a standard licensure and approval of a biologic, a category that includes vaccines. Dr. Marks also expressed confidence in the timeline for the EUA process, citing that the majority of adverse events that are related to vaccination occur within the first six weeks after the vaccine was administered. Most of the vaccines currently under study require two doses, which are typically given three to four weeks apart, and this critical six-week period would occur after the second dose has been administered. Other members of the panel discussion added that an EUA may not mean that a vaccine will be imminently available for delivery. Manufacturing, distribution, and delivery are equally important topics that are not covered in this piece.
Many observers have also begun to speculate how a vaccine would be made available once or if an EUA is issued. Some have suggested that it would be most appropriate for highest risk people first, such as front-line health care workers and nursing home residents. Whether this type of staged rollout will be part of an EUA, or whether prioritization handled by other authorities such as the CDC, or both, remains to be seen.
Selected phase 3 trials recruiting or underway:
| Developer | Vaccine name | Start date | Primary completion | Study completion | Participants Age in years | Site |
|---|---|---|---|---|---|---|
| AztraZeneca | AZD1222 | 8/17/20 (e) | 12/2/20 (e) | 10/5/22 (e) | 30,000 people Age: ≥ 18 | U.S. |
| Moderna | mRNA-1273 | 7/27/20 (a) | 10/27/22 (e) | 10/27/22 (e) | 30,000 people Age: ≥ 18 | U.S. |
| Pfizer/BioNT | BNT162 | 4/29/20 (a) | 4/16/21 (e) | 11/11/22 (e) | 29,481 people Age: 18-85 | U.S. |
| AstraZeneca | ChAdOx1 (AZD1222) | 5/28/20 (a)
| 8/1/21 (e)
| 8/1/21 (e)
| 12,330 people Age: 5-12, ≥ 18 | U.K. |
a = actual; e = estimated
Primary completion: The date which the last participant in a clinical study was examined or received an intervention to collect final data for the primary outcome measure.
Study completion: The date which the last participant in a clinical study was examined or received an intervention/treatment to collect final data for the primary outcome measure, secondary outcome measure, and adverse events.
International regulatory bodies in Europe, Russia and China all have mechanisms in place to allow for conditional or early approval of a vaccine. China and Russia have both already given early approval for vaccines developed in their countries. If a candidate vaccine in the U.S. received EUA, the FDA would need to determine who would be eligible to receive it under this type of authorization. It is possible that the authorization could be limited to people at high risk of disease and/or exposure such as the elderly, or health care providers. It is also possible that it could be authorized for the general public. For the AstraZeneca vaccine, phase 3 trials in the US are being conducted for adults only. Children had been enrolled in the U.K. sites of the trial, and data from international trials may be considered adequate for EUA.
Now more than ever, as public trust in the federal government is being undermined, health experts warn that missteps from the CDC and FDA are increasingly worrisome. At a time when vaccine hesitancy threatens the chance for a vaccine to pave a path towards a new normal, any shortcuts, cut corners, or hasty and politicized decisions around approval and use must be avoided. Experts have sounded alarms and warnings regarding a vaccine EUA, but also spoken of their confidence that there may be adequate data to support such a designation in the near future. A diversity of efforts, and fully transparent information, will be necessary to gain the confidence of diverse populations in the U.S..
FAQ
How do we track deaths from COVID-19?
Vital statistics, also called vital records, refer to essential data gathered from important or defining events in a population, which typically includes information on births, deaths, marriages and divorce. Vital statistics serve an important function by allowing authorities to record, track and analyze information related to these events on a population level and to identify trends which may be of interest on a multitude of levels. In public health, aggregated data on causes of death are of interest because they can reveal patterns in the burden of major diseases that can be used to design, implement and monitor health programming and interventions to improve health. This can be especially helpful when there are changes in patterns of disease, such as with the opioid epidemic, or when there are novel and urgent health threats, such as the COVID-19 pandemic.
In the U.S., the Centers for Disease Control and Prevention (CDC) oversees the collection, analysis and reporting of this data through the National Center for Health Statistics, although the recording of vital events falls under the jurisdiction of individual states. Mortality data including cause of death, has been systematically recorded in the U.S. by all 50 states since 1933, with revisions to allow for reporting of race, adjusting for an aging population, and to allow for electronic reporting. This information is included in a death certificate: a legal document that is issued in the event of any death, and requires attestation by a physician or coroner to validate the identity of the deceased and the cause of death.
States and jurisdictions collect and certify data on deaths then send the information to the CDC on a regular basis, and sometimes as close to real-time as possible. Although data on cause of death is analyzed regularly and reported annually, there are times when its rapid and timely analysis are critical to understand urgent public health threats. Mortality surveillance has been an integral part of tracking the impact of the COVID-19 pandemic and enhancing public health interventions and health care system responses. In order for these data to be meaningful, they need to be reported quickly and accurately. Some states have had difficulties with their death reporting, where official death counts have varied from what is being tracked at local and county levels. There are several points in the process of documenting cause of death that can lead to inaccuracies, including when forms are not filled out accurately, when forms are not filled out completely, when information on forms is not coded properly (taking the words written by physicians and coroners and converting to diagnosis codes used by data analysts) and when there are issues with transmitting information from localities to states to federal authorities. To aggregate this data in a more timely manner, some independent entities such as universities, newspapers and online initiatives track tallies of deaths daily at the county and state level, where data may come from death certificates, but may also come from reporting hospitals. Some sites scour several data sources including state dashboards, official statements and hospital announcements to quantify deaths daily as close to real time as possible. Official certified deaths reported to the CDC may take longer to aggregate, collate and analyze.
Cause-of-death reporting in the U.S. allows for conditions to be reported as the underlying cause of death as well as the immediate, or direct, cause of death. For example, if a person most immediately dies of heart or lung failure, but this heart or lung failure was brought on by a severe pneumonia from COVID-19, then COVID-19 will be listed in a sequence of events that leads to a death but starts with COVID-19. In fact, it is expected that there would be multiple events or causes of death listed on a carefully completed death certificate. Similarly, though someone may have severe brain swelling as the direct or immediate cause of death, this may have been secondary to bleeding in the brain after a fall or trauma—the underlying cause of death. Someone who has a large blood clot in the lungs, a pulmonary embolism, may have developed this problem due to a cancer—the underlying cause of death. For this reason, multiple problems may be included in the section of the death certificate that deals with the conditions that directly lead to death, and those that are underlying conditions. In addition, death certificates can include information about other health problems that contribute to someone’s death such as diabetes or obesity. The CDC provides instructional videos and other content for people responsible for completing death certificates to ensure certificates are completed as accurately as possible. It also has definitions for confirmed and probable deaths from COVID-19. A death is caused by COVID-19 whether it is reported as a direct cause of death or underlying cause of death.
Example of the “Cause of Death” section in a death certificate for someone deceased from COVID-19

Recently, there was confusion about a statistic used in a CDC publication. This was due, in part, to the lack of understanding about how deaths are recorded and tracked in the U.S. in general, as well as variation in the level of detail and accuracy provided on some death certificates. By expert accounts, the current tally of deaths attributed to COVID-19 is not an overestimate, but in fact likely a significant underestimate. The true toll of the pandemic may not be realized for years, but accurate and timely reporting through death certificates now can help to better understand the pandemic and make efforts to reduce its impacts.
Weekly Research Highlights
Community Outbreak Investigation of SARS-CoV-2 Transmission Among Bus Riders in Eastern China
(JAMA, September 1, 2020)
- This outbreak investigation identified a total of 31 confirmed COVID-19 cases among 300 participants attending a 150 minute outdoor worship service. Twenty-four of the cases (including the apparent index case patient) occurred among 68 passengers who had traveled aboard one of two buses arranged to bring people to and from the event. No cases occurred among the 60 passengers in the other bus and seven cases were identified among the remaining 172 participants who traveled to the event by other means.
- On the 100 minute round trip, passengers remained in the same seats throughout both legs of the journey and did not move about the cabin. The passenger later identified as the probable index case sat in a central location and was asymptomatic until after the return trip. The remarkably high attack rate (34.3%, 95% Confidence Interval [95% CI]: 24.1 to 46.3) among passengers on the implicated bus was 42.4% (95% CI: 27.2, 59.2) for those seated near the index case patient and 26.5% (95% CI: 14.4, 43.3) for passengers seated at least three rows away.
- The authors conclude that the lack of a statistically significant difference in risk based on distance from the index case patient suggests airborne transmission may have played a role. They note that the air conditioning units were operating in the recirculation mode throughout and only some of the windows could have been opened. Awareness of COVID-19 was low at the time and none of the passengers wore face masks nor were alerted to maintain physical distance.
- This is a well characterized outbreak investigation of a superspreader event strongly linked to exposure on the bus. Although the findings suggest the possibility of airborne transmission conveying viral aerosols over long distances, it is also likely that larger respiratory droplets may have been propelled by ventilation or transmitted during close contact among passengers before or after travel and while boarding the bus. We have previously described how difficult it can be to pinpoint the relative contributions of aerosol and droplet transmission for respiratory infections.

FIGURE: Seating chart on bus implicated in transmission of 24 cases of COVID-19 on January 19, 2020.
Saliva or Nasopharyngeal Swab Specimens for Detection of SARS-CoV-2
NEJM, August 28, 2020
- 70 patients hospitalized with COVID-19 were retested for SARS-COV-2 by quantitative reverse-transcriptase polymerase chain reaction (RT-qPCR) performed on pairs of samples: saliva samples collected by the patients themselves and nasopharyngeal samples collected at the same time point by health care workers. In addition, 495 asymptomatic health care workers provided self-collected saliva and self-collected nasopharyngeal samples for SARS-CoV-2 testing.
- Among hospitalized patients, more copies of SARS-CoV-2 RNA were detected in saliva specimens than in nasopharyngeal specimens, there was less variation in SARS-CoV-2 RNA levels in saliva specimens than in nasopharyngeal specimens, and it was more common to obtain a negative result followed by a positive result in a series of nasopharyngeal specimens from a single patient (3 instances) than in a series of saliva specimens from a single patient (1 instance). At 1 to 5 days after diagnosis, 81% (95% CI, 71 to 96) of the saliva specimens were positive, as compared with 71% (95% CI, 67 to 94) of the nasopharyngeal specimens.
- Among asymptomatic health care workers, SARS-CoV-2 RNA was detected in the saliva of 13 people (those results were later confirmed by repeat nasopharyngeal testing). Nine of those 13 people had submitted paired nasopharyngeal samples, and seven of those samples tested negative for SARS-CoV-2.
- The statistical significance of the observed differences is not clear. The population of symptomatic patients was restricted to those already diagnosed with COVID-19 on nasopharyngeal testing and hospitalized.
Humoral Immune Response to SARS-CoV-2 in Iceland
NEJM, September 1, 2020
- Over a four-month period, SARS-CoV-2 antibodies were measured in up to 30,576 Icelanders. Some had tested PCR-positive (some were currently hospitalized and some had recovered) and some had either never been tested or had tested PCR-negative (some had been in quarantine and some had not).
- Of 1,215 people who had recovered from COVID-19, 1,107 (91.1%; 95% confidence interval [CI], 89.4 to 92.6) were seropositive. Over 90% of those who tested positive for IgG antibodies remained seropositive 120 days after diagnosis, with no decrease of IgG antibody levels. Antibody levels were higher among those who were older, those with a higher body mass index and those with more severe COVID-19. Antibody levels were lower among smokers and those who use anti-inflammatory medications.
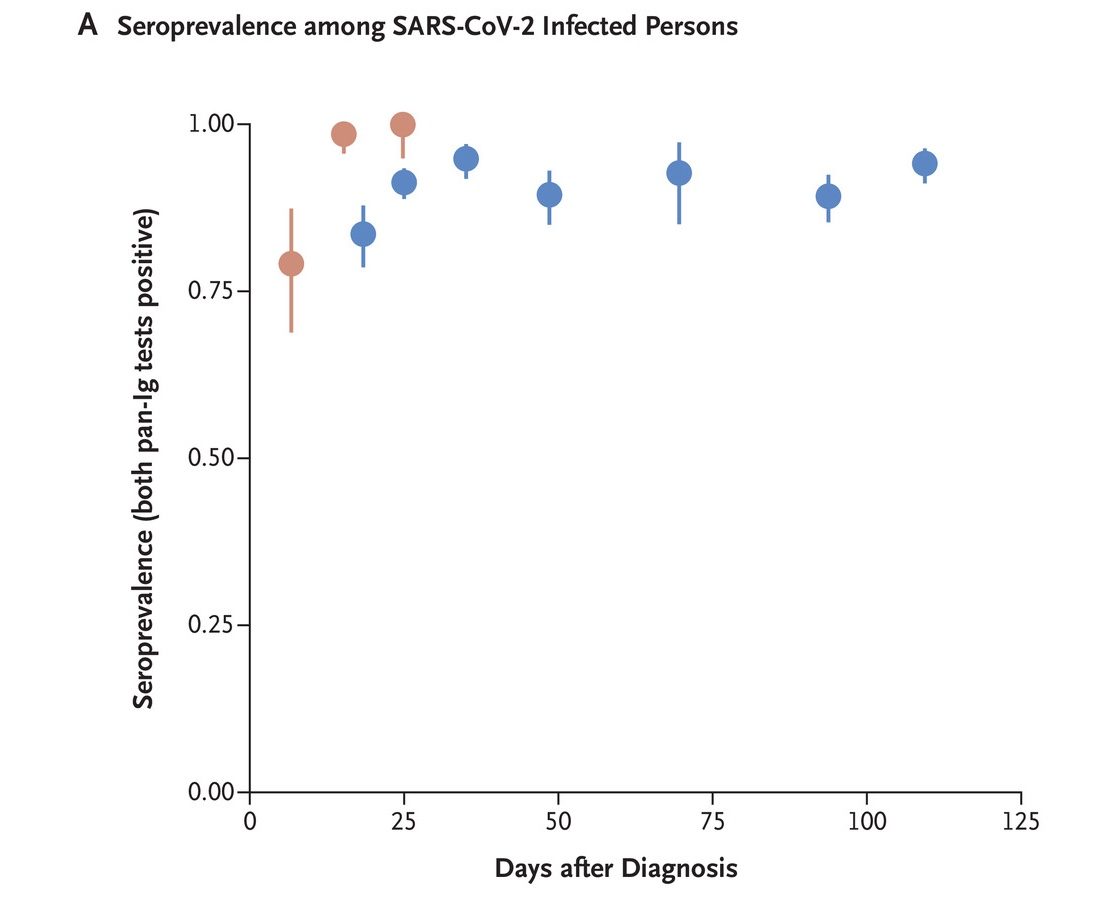
Red denotes the percentage of positive samples among hospitalized people (249 samples from 48 persons), and blue denotes the percentage of positive samples among recovered people (1,853 samples from 1,215 people).
- Approximately 56% of all SARS-CoV-2 infections were diagnosed by PCR. Of total infections, 14% were in quarantined people who were not PCR-positive (defined as never been tested or tested negative), and 30% were in people who had not been in quarantine and had not been PCR-positive.
- Among 4,222 quarantined people who had not tested PCR-positive, 97 (2.3%; 95% CI, 1.9 to 2.8) were seropositive; those with household exposure were 5.2 (95% CI, 3.3 to 8.0) times more likely to be seropositive than others.
- Considering the results of PCR testing alone, the incidence of SARS-CoV-2 infection in Iceland was 0.5%; considering both PCR testing and antibody testing, the incidence of infection in Iceland was 0.9% (95% CI, 0.8 to 0.9). Considering the results of PCR testing alone, the infection fatality rate was 0.6%; considering both PCR and antibody testing, the infection fatality rate was 0.3% (95% CI, 0.2 to 0.6).
- Results were obtained from a relatively homogenous population in a specific geographical region. Further studies will be necessary to determine the longevity of SARS-CoV-2 antibodies in other populations.
Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis
JAMA, September 2, 2020
Main message: There have been multiple studies on synthetic corticosteroids for the treatment of COVID-19. When interim results from the U.K.-based RECOVERY trial on a number of potential COVID-19 treatments showed a significant reduction in mortality among COVID-19 patients who had been given the steroid dexamethasone, other steroid trials were suspended because withholding steroids could no longer be justified. This meta-analysis of existing data on steroid treatment for COVID-19 patients upholds the findings of the RECOVERY trial: compared with usual care or placebo, steroids were associated with lower 28-day all-cause mortality among critically ill COVID-19 patients.
- Data from trials that randomly assigned critically ill COVID-19 patients to a steroid group and to a non-steroid group were eligible for inclusion. The primary outcome was all-cause mortality 28 days after randomization; the secondary outcome was serious adverse events.
- Seven trials were included in the analysis. Different trials administered different types (dexamethasone, methylprednisolone and hydrocortisone) and doses of steroids. A total of 1,703 patients from countries on five continents were randomized: 678 to receive steroids and 1,025 to receive usual care or placebo. The median age was 60 years and 488 patients (29%) were women. The proportion of patients receiving concurrent treatment with other potential antiviral agents varied between trials.
- There were 222 deaths among 678 patients randomized to receive steroids and 425 deaths among 1,025 patients randomized to usual care or placebo. The summary odds ratio (OR) of 28-day mortality was 0.66 (95% CI, 0.53-0.82). Among the six trials that reported serious adverse events, there were similar numbers of severe adverse events among patients receiving steroids and patients receiving usual care or placebo.
- Subgroup analysis showed that both dexamethasone and hydrocortisone, but not methylprednisolone, conferred similar mortality benefits. For mechanically ventilated patients, the OR was 0.69 (95% CI, 0.55-0.86); for unventilated patients, the OR was 0.41 (95% CI, 0.19-0.88). Similar mortality benefits were found for both male and female patients and for both older and younger patients.
- The study was underpowered to assess optimal steroid dosing, and treatment duration could not be assessed. Only adults in high-income settings were included. Mortality data beyond 28 days were not available. Although it appears that steroids may be more beneficial in those not mechanically ventilated, only 144 patients were not receiving mechanical ventilation at randomization.
Transmission Dynamics of COVID-19 Outbreaks Associated with Child Care Facilities — Salt Lake City, Utah, April–July 2020
(MMWR, early release September 11, 2020)
- Researchers analyzed data from three child care facility outbreaks in Utah from April to July, 2020 to determine attack rates and transmission patterns. Investigators identified a total of 184 people epidemiologically linked to the affected child care facilities, 110 of which were children, and 83 of which were non-facility contacts (e.g., siblings or parents of children attending child care facilities).
- Of the people known to have an epidemiologic link to the child care facilities, 31 confirmed and seven probable cases of COVID-19 were identified. Most of the cases (71%) were among staff and attendees of the facilities, however nine confirmed cases and seven probable cases occurred among contacts, including one case in a parent who required hospitalization. Transmission from an asymptomatic child to a contact was documented in at least two instances. The index case at all three facilities was in an adult staff member.
- Mitigation strategies including the use of masks among all staff and attendees over two years old, excellent hygiene, and other practices such as stable staff/attendee cohorts without mixing, can be critical to keep these types of settings safe and open. Once a case is identified, timely testing and tracing of contacts, and other steps such as quarantining, are critical to control outbreaks and limit ongoing transmission.
Suggested citation: Cash-Goldwasser S, Kardooni S, Cobb L, Bochner A, Bradford E and Shahpar C. In-Depth COVID-19 Science Review August 29 – September 11, 2020. Resolve to Save Lives. 2020 September 15. Available from https://preventepidemics.org/covid19/science/review/



