



How genomic surveillance helps us understand SARS-CoV-2
Why do we use genomic surveillance?
Genomic surveillance is widely used in public health. In the United States, the PulseNet Program provides routine genomic surveillance of foodborne pathogens (e.g., E. coli and Salmonella). By helping to identify 1,500 state or local outbreaks and 250 multistate clusters of foodborne illness each year, the program has been integral to improving food safety. During the 2014-2016 Ebola outbreak in West Africa, sequencing data in combination with epidemiologic data clarified whether outbreaks were new or continuing, and showed that the Ebola virus could be transmitted sexually. As a result, the World Health Organization (WHO) recommended semen testing for male Ebola survivors and that people practice safer sex. Genomic surveillance can also inform clinical decision-making. For example, genomic surveillance of malaria parasites can detect the spread of drug resistance and inform approaches to prevention and treatment.
During the COVID-19 pandemic, genomic surveillance has helped scientists make important discoveries about the novel SARS-CoV-2 virus. WHO identified several public health objectives of SARS-CoV-2 genomic sequencing, establishing the activities that can be achieved with limited, occasional sequencing and the activities that require sustained, coordinated and more comprehensive genomic surveillance programs (Table).

Source: World Health Organization
The fulfillment of such objectives requires careful planning and coordination of genomic and other surveillance systems, as well as enormous investments in technical, logistical, human and financial resources. The basis of any genomic surveillance system is the technology needed to detect genetic changes. To make sense of the changes detected and inform public health action, a number of other tools and resources are required. These include the data tools and scientific knowledge to analyze and interpret what detected changes mean as well as the infrastructure to do sufficient monitoring over time and to conduct epidemiologic investigations.
How does the SARS-CoV-2 genome change and what could those changes mean?
The genetic code consists of a series of chemical building blocks that encode for the proteins that make up the structure of the organism. These building blocks are linked into strands to form either DNA or RNA. Proteins called polymerases transcribe the DNA or RNA strands, creating copies from template strands. All polymerases occasionally make errors, substituting one chemical building block for another when transcribing a new genome instead of perfectly replicating the template; this is one way in which mutations may occur. Coronaviruses have RNA genomes, and RNA polymerases are particularly error-prone. Some RNA viruses mutate especially frequently and have higher mutation rates than coronaviruses. For example, HIV lacks a proofreading enzyme that corrects replication mistakes and influenza viruses have segmented genomes with parts that are regularly mixed and matched. Genetic mutations, including those that arise in SARS-CoV-2, may or may not have significant biological implications. Over the course of the COVID-19 pandemic, researchers have documented many mutations in SARS-CoV-2 genomes, but only a small fraction have been shown to be biologically important.
Many mutations will have no impact on the function of the virus and some may reduce its viability. Virions (individual viral particles) with mutations that offer no advantage—and especially those that have disadvantageous mutations—will not outcompete existing viruses. Alternatively, virions with mutations that increase viability may outcompete other virions, increasing the prevalence of those mutations over time. Mutations in the SARS-CoV-2 gene encoding for the spike (S) protein may be biologically significant because the S protein is the part of the virus that initiates infection by binding to the ACE-2 human cell receptor. As one example, in early March 2020, researchers noted the increasing prevalence of SARS-CoV-2 isolates with a mutation in the S gene that resulted in a change in the S protein (at position 614, the protein building block notated as “D” had changed to the building block notated as “G”—hence a “D614G” substitution). Over time, SARS-CoV-2 variants with the D614G substitution became the dominant form of SARS-CoV-2 around the world. Researchers have hypothesized that the D614G substitution resulted in the S protein binding more easily with the ACE-2 receptor, contributing to the generation of larger amounts of virus and thus to greater transmissibility.
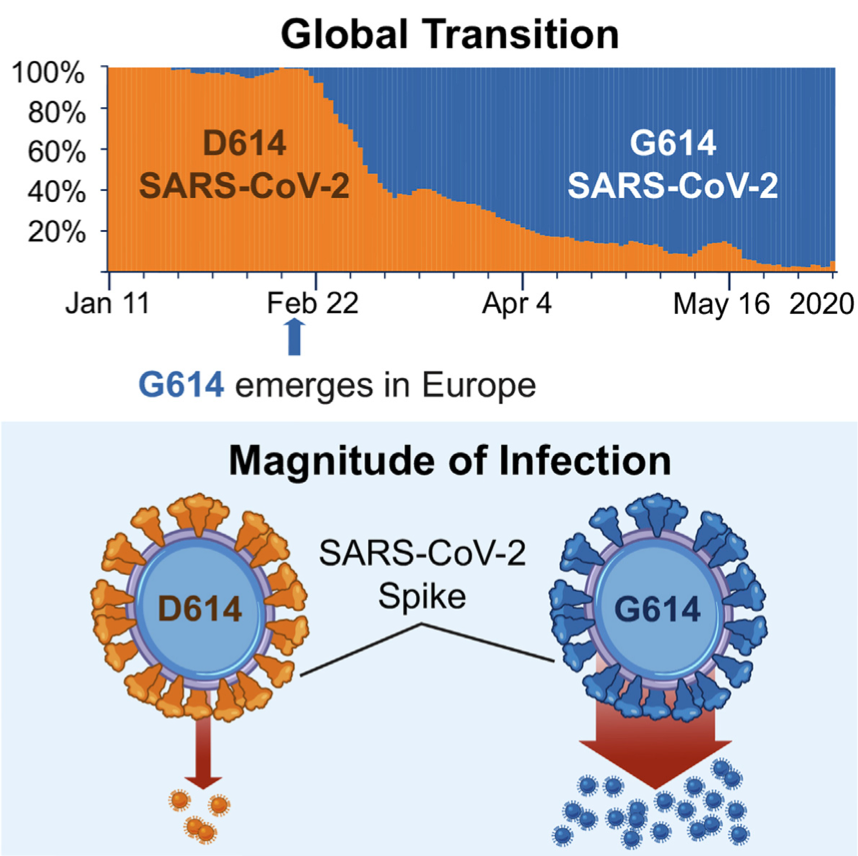
Source: Cell.com
How do we monitor changes in the SARS-CoV-2 genome?
Genomic surveillance is the monitoring of genomic sequencing data on a pathogen. Genomic surveillance enables scientists to monitor changes in the frequency of mutations, such as the D614G mutation, over time, and to investigate how these changes might affect health. In order to conduct genomic surveillance it is necessary to perform genetic sequencing. Then those data must be processed, stored, combined with other types of data, analyzed and interpreted to meaningfully inform public health action.
Genomic sequencing is the process of determining the order of chemical building blocks within a genetic code. Genomic sequencing of pathogens is performed in a laboratory using a biological sample obtained directly from a patient or an isolate from a patient that has been grown in the lab (e.g., a viral isolate grown from cell culture). After the genetic material is extracted from the sample, a machine is used to “read” the chemical building blocks that make up the genetic code. The resulting sequences are represented as a series of letters, with each letter representing a different building block (e.g., ATTGGAC).
Sequencing data produced by the machine needs to be processed. Sequencing machines typically generate a large number of “reads”—or fragments of genetic sequence—that must be trimmed, aligned and then combined to generate a consensus sequence based on the best guess of the letter at each position in the genome. Extraneous genetic sequences that are not from the intended pathogen also need to be removed (e.g., human genetic material must be removed from samples that contain a mix of human and pathogen genetic material).
Once processed, consensus sequence data are often uploaded to public databases to guide research and public health response around the world. The Global Initiative on Sharing All Influenza Data (GISAID) is one such database. This database serves as a repository for genomic, clinical and epidemiological data, to help researchers understand how viruses evolve and spread during outbreaks. GISAID was initially developed for influenza surveillance but has been used extensively for SARS-CoV-2. The initial SARS-CoV-2 genome sequences from China were posted to GISAID and many SARS-CoV-2 research efforts continue to use this platform.
Genomic sequence data are of limited utility without accompanying “metadata.” Metadata include both information about the process used to derive the sequence (i.e. laboratory and data analytic tools used) and information about the sample and patient (e.g. date of collection, patient location). The latter is particularly important for epidemiologic investigation and responding to an outbreak. Metadata can also include patient demographics (age, sex), travel history and contacts with other cases, along with clinical information such as types of symptoms, whether the patient was hospitalized, history of COVID-19 vaccination, whether they have comorbidities and the clinical outcome of illness (e.g., fatal or not) and how many of their contacts became infected.
How do we make sense of the SARS-CoV-2 genetic changes detected by genomic surveillance?
One type of analysis that can be performed using genomic surveillance data is phylogenetic analysis, which uses genetic sequences to infer evolutionary relationships. In the case of SARS-CoV-2, genomic sequence data from multiple isolates are used to hypothesize the order in which different SARS-CoV-2 mutations arose in a population over time and estimate how different variants are related through common ancestors. The data are commonly visualized as a “phylogenetic tree,” with the ancestor sequence as the root of the tree and new mutations appearing as branches.
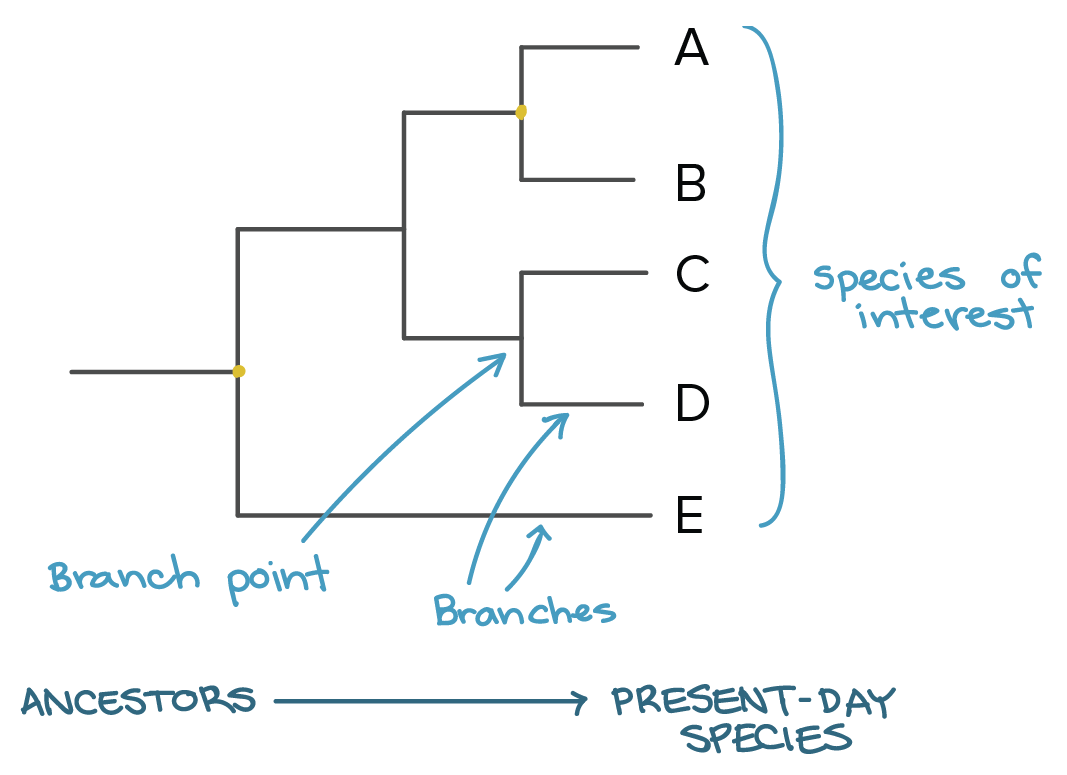
Source: Khan Academy
Viral genetic diversity may be categorized into distinct “clades” or “lineages,” each of which includes a founding variant and its descendants, with each clade corresponding to a branch or cluster of branches on a phylogenetic tree. In the phylogenetic tree below, SARS-CoV-2 isolates are represented as dots grouped on branches according to their genetic similarity, with branches grouped by the founding variant clade. Whereas genomic surveillance provides core genetic data for a phylogenetic tree, associated metadata on geographic location and timing of clinical sample collection is necessary to construct the tree.
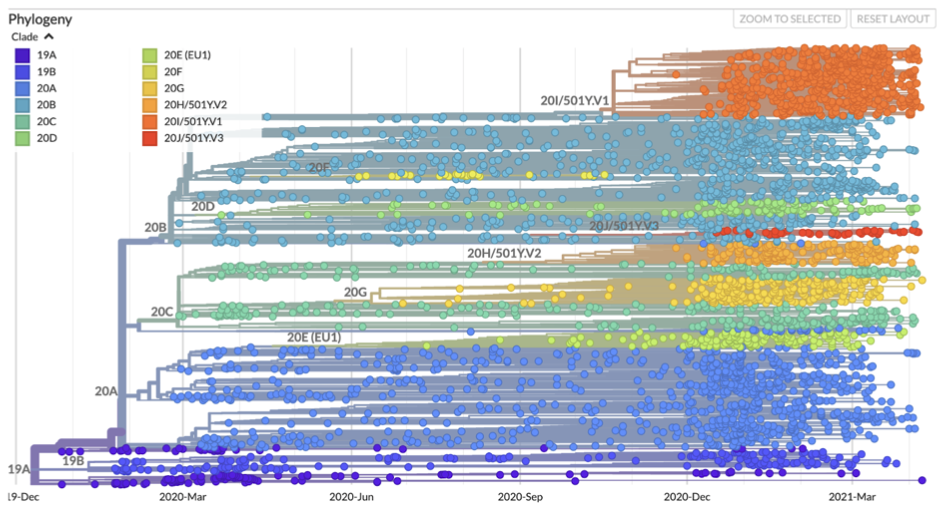
Source: Nextstrain.org
Of note, there is not yet a consensus on SARS-CoV-2 variant nomenclature. The phylogenetic tree above shows Nexstrain nomenclature (clades are named for the year in which they emerged and assigned a letter—e.g., 19A, 19B, 20A), as well as nomenclature based on the location of a signature mutation (a switch to the “Y” building block in position 501 is denoted as 501Y mutation). Another proposed nomenclature system, the Pango nomenclature system, assigns lineages ascending letters and numbers (e.g. B.1.1.2, B.1.1.3).
Regardless of the nomenclature used, different variants have varying public health implications. The U.S. Centers for Disease Control and Prevention (CDC) uses the results of clinical, epidemiologic and laboratory studies to classify variants by their potential impact on pandemic countermeasures. A variant may be classified as a “Variant of Interest,” a “Variant of Concern” or a “Variant of High Consequence” if it has one or more of the attributes listed in the columns of the table below.
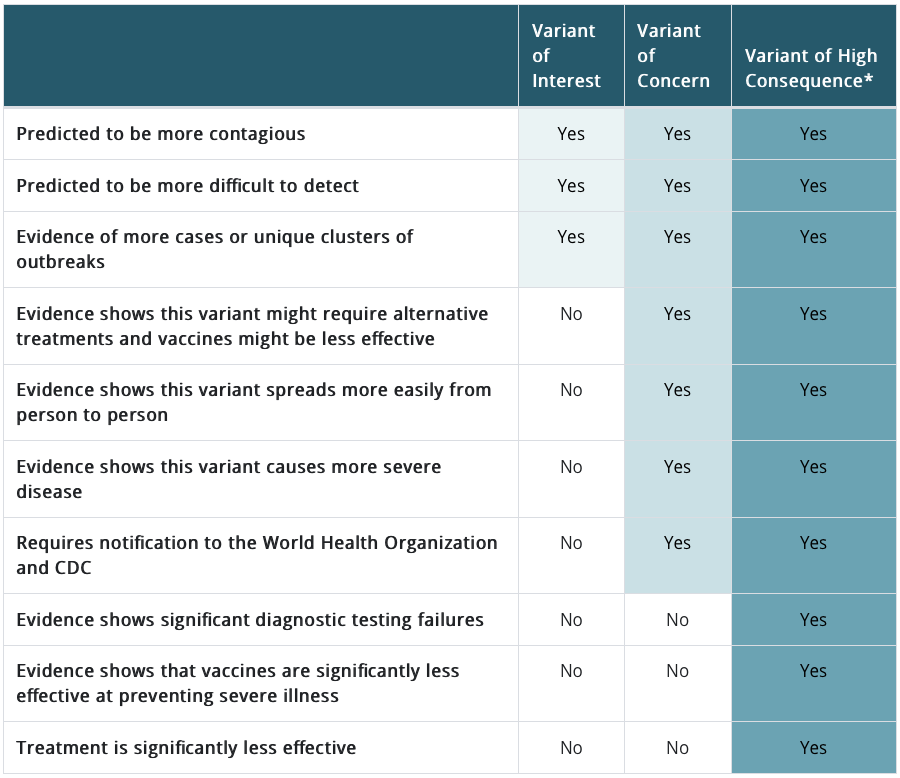
Source: CDC
Currently, the CDC has identified three variants of interest and five variants of concern circulating in the United States. For example, the Pango lineage P.2 variant is considered a variant of interest because it has mutations that may confer resistance to neutralizing antibodies, whereas the Pango lineage B.1.1.7 variant is a variant of concern because evidence shows it is more transmissible and more deadly. These classifications illustrate that while genomic surveillance is critical to identify the emergence and spread of new variants, the analysis and interpretation of genomic data also requires other types of information from epidemiological, laboratory and clinical studies.
What are some examples of how genomic surveillance has been used during the COVID-19 pandemic?
1) Identify the novel pathogen
The rapid response team deployed to Wuhan, China in December 2020 to investigate a cluster of patients with pneumonia of unknown cause used genomic sequencing to identify the SARS-CoV-2 virus. Scientists applied a technique called unbiased high-throughput sequencing to lower respiratory samples from three patients hospitalized at Wuhan Jinyintan Hospital. This approach sequences all of the genetic material present in the samples, facilitating the discovery of completely new pathogens such as SARS-CoV-2. The scientists submitted the novel sequence to GISAID. In January 2020, comparing sequences of 12 SARS-CoV-2 isolates from China helped scientists identify that there was likely human-to-human transmission of the virus.
2) Track transmission and inform disease control measures
Integrated with epidemiologic data, SARS-CoV-2 genomic data have been used to identify settings where transmission is occurring, map spatiotemporal patterns in disease spread and inform disease control. A study of genomic sequencing data from 772 COVID-19 cases in Boston from March to May 2020 helped demonstrate the importance of superspreading events in COVID-19 transmission. Epidemiologic investigation identified at least 100 cases linked to a business conference in Boston in late February. Sequencing data were available for 28 of these cases and specific mutations that characterized cases associated with the conference were identified. Researchers looked for these mutations in genomic sequences available from across the U.S. and estimated that the superspreading event at the Boston conference was linked to community spread that gave rise to as many as 50,000 reported cases across multiple states by the end of May 2020, and potentially to more than 300,000 cases by Nov. 1, 2020. More recently, genomic sequencing helped demonstrate that insufficient quarantine of newly transferred incarcerated persons sparked a COVID-19 outbreak in a Wisconsin prison. Findings like these underscore the importance of policies to limit large indoor gatherings and ensure adequate resources for testing and quarantine in high-risk settings.
Genomic surveillance also informed policy-level decisions on pandemic mitigation measures in Victoria, Australia, where sequencing data were available for 80% of cases. Public health officials used SARS-CoV-2 genomic data to identify links between cases with no apparent epidemiological connection. For example, four case clusters and several unlinked cases, all from the same metropolitan area, were found to form one genetic cluster. This evidence of community transmission (which was not apparent from contact tracing information) supported implementation of community-level social restrictions. After implementation of those restrictions, no further cases were associated with the genetic cluster, which suggests that transmission was effectively interrupted.
In addition, genomic sequencing has been used to track the geographic spread of SARS-CoV-2. This includes determining whether new cases are descended from prior cases detected in the country, which indicates ongoing transmission—or if the new cases are genetically different, which indicates a novel incident of disease being imported from a different geographic region. For example, public health officials in Minnesota used genomic surveillance to detect the first instance of the P.1 variant in the U.S. on Jan. 25, 2021, just 19 days after the variant was first isolated in travelers returning to Japan from Brazil. This reflected a pattern of ongoing, rapid, international spread of COVID-19.
3) Inform prevention, diagnostic and treatment strategies
An increasingly important function of genomic surveillance is to support vaccine development. A prime example is the development of seasonal influenza vaccines. Due to the high mutation rate of influenza viruses, a new seasonal influenza vaccine must be produced each year. Throughout the year, health care centers and labs conduct influenza surveillance by collecting samples from patients and testing them for influenza. Each year, the results of genomic surveillance on these isolates inform the development of that year’s seasonal influenza vaccines.
The first COVID-19 vaccines authorized for use in the United States, mRNA vaccines, were designed using SARS-CoV-2 genomic data that was obtained from early COVID-19 patients in China and uploaded to GISAID. New variants of concern, including the B.1.1.7, B.1.351 and P.1 variants, emerged after the development of the COVID-19 vaccines that are currently in use. Most of these vaccines, including the Johnson & Johnson, Moderna, Oxford-AstraZeneca and Pfizer-BioNTech vaccines, are designed to induce immune responses to the S protein. There is concern that these vaccines may be less protective against variants with mutations in the S gene. So far, evidence from antibody neutralization studies in the laboratory, vaccine efficacy trials and real-world vaccine effectiveness studies, suggest that several of the aforementioned vaccines offer excellent protection against severe disease caused by the B.1.1.7 and B.1.351 variants, although further investigation is needed, and data on the P.1 variant are lacking. Some breakthrough COVID-19 cases (occurrences of infection despite vaccination) are expected to occur, including infections caused by SARS-CoV-2 variants that do not have new mutations. It is critical that genomic surveillance data be linked to data on these breakthrough cases, so that the reasons for vaccine failure are explored and adjustments to vaccine formulations or schedules can be made if necessary. Efforts are underway to develop and test booster vaccines that may offer better protection against specific SARS-CoV-2 variants.
An additional purpose of genomic surveillance is to detect variants that may reduce the accuracy of diagnostic tests that detect SARS-CoV-2 genes (PCR tests) or proteins (antigen tests). For example, variants may not be detected by PCR tests that target the mutated gene. Indeed, when the B.1.1.7 variant emerged in the U.K., it was observed that PCR tests targeting both the S and other genes delivered a negative result for the S gene target and a positive result for the alternative target, a phenomenon known as “S gene target failure.” Fortunately, PCR tests that target only the S gene are not widely used in clinical and public health settings; rather, most authorized PCR tests target multiple genes in part to avoid the failure of a single target if a mutation has occurred. In this case, S gene target failure in a multi-target PCR test, which suggests the presence of a variant with an S gene mutation, has served as a useful surveillance tool. Researchers have used rates of S gene target failure to estimate that the B.1.1.7 is more transmissible and likely more deadly than other variants.
Genetic mutations may also have therapeutic implications. There are numerous examples of pathogens for which specific genetic mutations have been linked to treatment successes and failures. Viral sequencing data helps guide the selection of HIV treatment regimens. Genetic testing can be used to guide selection of treatment for certain bacterial infections, such as tuberculosis and gonorrhea, that require immediate treatment but may be drug-resistant and can be difficult or slow to grow and test in the lab. As we previously wrote, some of the S gene mutations of known SARS-CoV-2 variants have been associated with reduced neutralization by monoclonal antibody therapies and the antibodies in convalescent plasma. The clinical implications of these findings are not well described, and genomic surveillance will continue to inform research in this area. There may someday be a defined clinical role for SARS-CoV-2 genetic analysis to guide treatment decisions.
Sequencing data has also been used to confirm the occurrence of SARS-CoV-2 reinfection. For example, a 25-year old man in Nevada tested positive for COVID-19 in April and June 2020. Evidence that supported reinfection included genetic sequencing data that showed that the infections in April and June were caused by genetically distinct SARS-CoV-2 viruses. These and other instances of reinfection reinforce the importance of adherence to precautions to prevent COVID-19 such as mask-wearing and social distancing, and also underscore recommendations that previously infected individuals get vaccinated.
How much SARS-CoV-2 genomic surveillance is enough?
How much genomic sequencing is needed depends on the context and on the goals of genomic surveillance. More intensive sequencing is needed to establish chains of transmission within a local area compared to sequencing a smaller representative sample of cases for tracking global trends in pathogen evolution. A modeling study, which has not yet been peer-reviewed and was carried out by a biotechnology company that manufactures sequencing technology, suggested that sequencing isolates from 5% of COVID-19 cases is sufficient to detect novel variants with a prevalence of 0.1-1%. The World Health Organization has recommended sequencing at least 15 samples per week and up to 10% of positive samples from some sites. WHO recommends that sequenced samples should be representative of different population groups, geographies, spectrum of disease severity and time periods. It is particularly important to sequence samples from patients who have severe illness, from those who may have been re-infected, and from those who develop illness, and especially severe illness, after vaccination.
Currently, the extent of genomic sequencing of SARS-CoV-2 varies greatly across countries (Figure). According to data from the GISAID database, there are more than 600 sequences available per 1,000 cases in Iceland and approximately 475 sequences per 1,000 cases in Australia. In the United Kingdom, where the B.1.1.7 variant was first identified, there are about 75 sequences per 1,000 cases (7.5% of cases are sequenced). In contrast, sequencing is much more limited in most countries. In South Africa, there are 2 sequences available per 1,000 cases; only 0.3 sequences per 1,000 cases in Brazil; and 0.2 per 1,000 cases in Ethiopia. Limited availability of sequencing data has led to a call for building sequencing capacity throughout the African region within an existing network of laboratories in seven countries established by WHO and Africa Centres for Disease Control.

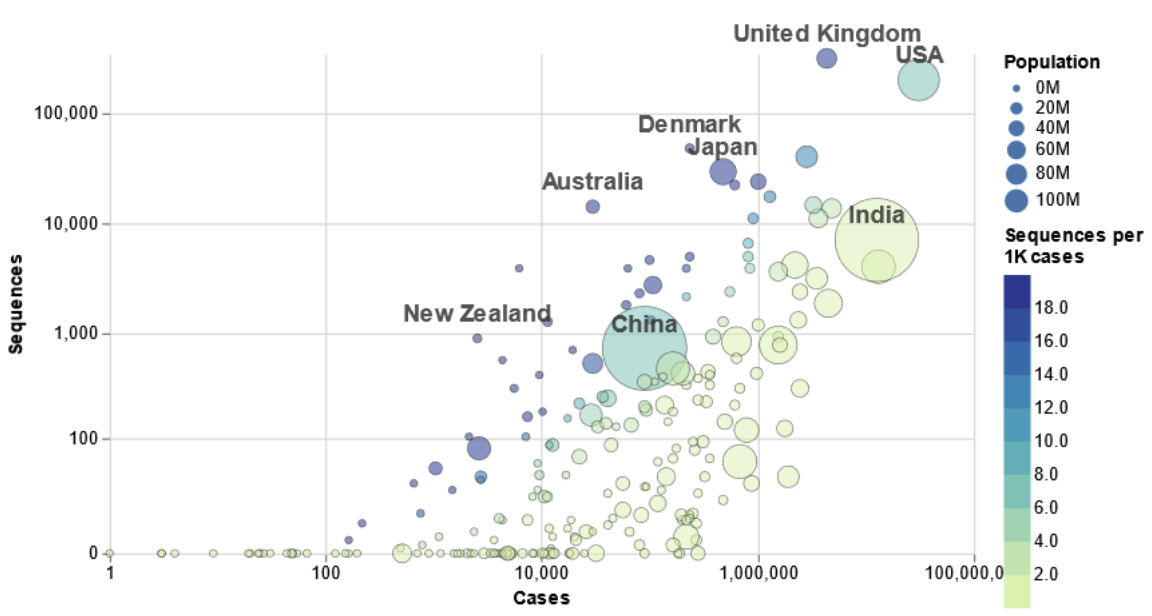
Source: COVID CG
In the United States, approximately 7 sequences are available per 1,000 cumulative cases, with rapid increases in the sequencing rate during the past four months. According to the U.S. Centers for Disease Control and Prevention (CDC), the number of SARS-CoV-2 sequences from the U.S. available on GISAID increased from 60,000 at the end of 2020 to over 240,000 as of early April (Figure). However, only 22 states have sequenced more than 1% of cumulative COVID-19 cases, and only Wyoming has sequencing data for >5% of cases. Genomic sequencing in the United States is carried out by the CDC, a network of public health laboratories, commercial laboratories and academic and medical centers. Data on the prevalence of SARS-CoV-2 lineages among isolates sequenced in the U.S. are available from the CDC.
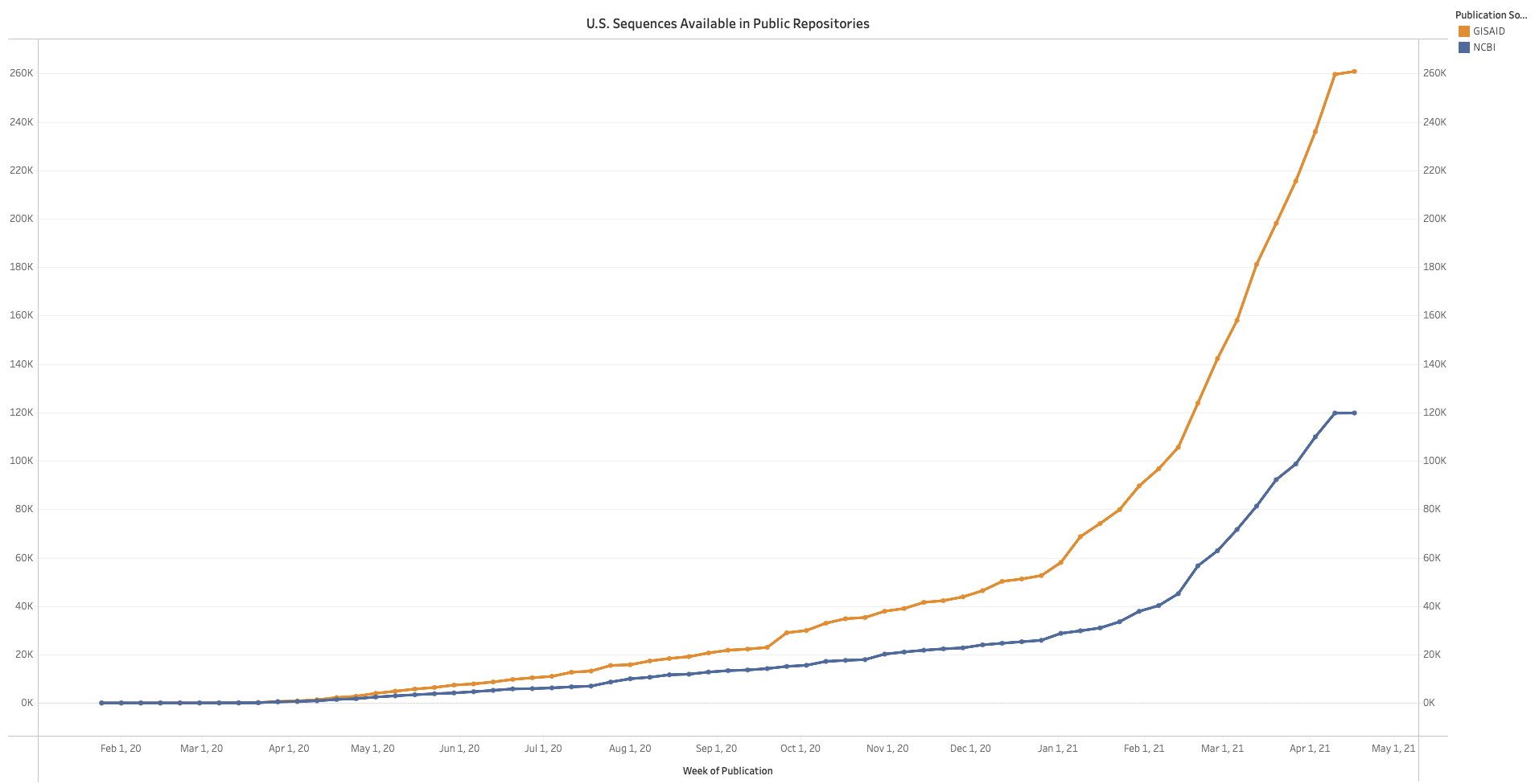
Source: CDC
What are the challenges and opportunities of genomic surveillance?
Over the past two decades, the price of sequencing a human genome dropped from USD $2.7 billion to $300, and sequence data from pathogens such as SARS-CoV-2 can be obtained for even less. Sequencing machines have also become smaller and faster. It is now feasible to perform sequencing at a large scale and with sufficient timeliness to inform public health action. Whereas early applications of genomic surveillance were often retrospective, these data are now available in real time, but to be most useful, even faster turnaround times —in the 1-2 day range—are needed. Genomic sequencing is now widely used in public health, with applications that include the identification and investigation of clusters or outbreaks of disease and the development and refinement of diagnostic tests, therapeutic agents and vaccines. The COVID-19 pandemic has demonstrated the tremendous potential value of genomic surveillance.
The pandemic has also exposed some persistent challenges of genomic surveillance, particularly in developing robust systems for collecting, processing, analyzing and interpreting genomic surveillance data, with linkages to epidemiological and clinical metadata. Effective genomic surveillance of infectious diseases is a global concern since pathogens do not respect borders, yet countries have vastly different resources to put toward developing and maintaining genomic surveillance systems. Although genomic data from SARS-CoV-2 isolates can be obtained for a relatively low price, the costs of setting up and conducting comprehensive genomic surveillance can be substantial. And it is not solely financial constraints that challenge the development of a robust genomic surveillance platform, but also constraints in human and technical resources. Without quality-assured reagents and laboratory expertise to help ensure that samples are stored and handled properly, the integrity of genetic material might be compromised. Processing of raw sequencing data requires bioinformatics tools and expertise, including access to high-performance computing power and digital storage space for large amounts of data; globally, the volume of SARS-CoV-2 data is now too large for many of the bioinformatics analysis tools that are currently available. Another challenge lies in linking genomic data to essential metadata. Whereas viral genomes are not protected by confidentiality regulations, some metadata are sensitive, protected health information. Therefore, it has been suggested that sensitive metadata be stored separately from viral genome sequences and non-sensitive metadata (e.g., state or country information, basic phenotypic information). Sensitive protected health information is used only for specific public health or clinical purposes by authorized individuals. Because sensitive patient data are stored separately from viral genetic sequences, linking viral sequences to clinical and epidemiological data has been a fundamental challenge that can limit the utility of genomic sequencing data.
Despite these challenges, genomic surveillance has increasingly become a valuable part of public health practice in recent decades. It can be a powerful tool when utilized as part of a comprehensive public health approach to preventing and controlling the spread of infectious diseases. There are already numerous examples of the ways in which genomic data on SARS-CoV-2 have informed the COVID-19 pandemic response. Across the globe, as resource challenges and questions about the implementation and role of SARS-CoV-2 genomic surveillance are addressed, and as activities are scaled appropriately to the objectives of surveillance in any given context, targeted genomic surveillance can be incredibly informative. We have already and will continue to improve mitigation measures when informed by genomic sequencing data from outbreaks, patients with severe illness, people who have been re-infected and people who develop COVID-19 after vaccination.
FAQ: Should pregnant people get a COVID-19 vaccine?
Pregnant people were not included in the original vaccine trials for the three vaccines currently authorized for use in the United States: Pfizer-BioNTech, Moderna and Johnson & Johnson. Although Pfizer began recruiting people between 24 and 34 weeks pregnant into a phase 2/3 study in February and will follow them through the first six months of their infants’ lives, this data will not be available this year. Because there are no long-term data on potential effects of the vaccine on infants whose mothers were vaccinated during pregnancy, pregnant people need to weigh the potential risks against the benefits of COVID-19 vaccination and make a decision in collaboration with their doctor. This FAQ will provide some tools to help inform that decision.
Recommendations at the time of authorization
Despite the lack of inclusion of pregnant women in the vaccine trials, the U.S. Food and Drug Administration’s Emergency Use Authorization allows for pregnant people to receive the approved vaccines. This is in line with guidance from the American College of Obstetrics and Gynecologists (ACOG), which recommends that people who are pregnant be offered the COVID-19 vaccine and the opportunity to make their own choice. The ACOG recommendation was made based on both what was known about vaccine safety at the time and the potential dangers of COVID-19 in pregnancy.
People who are pregnant are at an elevated risk for severe illness and death from COVID-19 compared to those who are not, and may be at risk of adverse pregnancy outcomes (e.g, preterm birth) if they contract COVID-19. Further, pregnant people who have comorbidities such as diabetes, hypertension or obesity may be at even higher risk of serious illness from COVID-19.
Data from pregnant people who have been vaccinated to date
Since the initial authorization, new data on the Moderna and Pfizer vaccines have emerged providing additional evidence that COVID-19 vaccinations in pregnancy are likely safe and effective (the Johnson and Johnson vaccine was released too recently to be included in the safety data). The CDC has been monitoring vaccine safety in pregnant women since December. As of mid-February, more than 30,000 pregnant people registered their vaccination with CDC, of whom 1,815 were enrolled in the v-safe pregnancy registry (232 have had a live birth). So far, the CDC has found that:
- There is no difference in side effects between pregnant and non-pregnant women vaccinated.
- Vaccinated women do not appear to have elevated rates of adverse pregnancy outcomes such as miscarriage and stillbirth, complications such as gestational diabetes or intrauterine growth restriction, or neonatal issues such as preterm birth or congenital abnormalities.
- While miscarriage was the most frequently reported adverse event, it occured less often among vaccinated women than in the general population.
A recent study published by Gray et al. in the American Journal of Obstetrics and Gynecology enrolled 84 pregnant, 34 lactating and 16 non-pregnant women at the time of vaccination. The study found that the mRNA vaccines (e.g., Moderna and Pfizer) work well in pregnant women, creating similar antibody responses to non-pregnant and lactating women. Furthermore, vaccines not only appear to protect pregnant people, but also to potentially protect their babies. Antibodies in pregnant women who are vaccinated appear to be transferred through the placenta to the baby (based on an assessment of antibodies in cord blood at delivery). They were also found in the breast milk of women vaccinated during lactation. Similar to the CDC safety data, the study also found no difference in side effects between pregnant and non-pregnant women.
There is currently no evidence that any vaccines, including COVID-19 vaccines, cause fertility problems. CDC recommends that people who are trying to become pregnant now or want to get pregnant in the future, may receive a COVID-19 vaccine.
Weekly Research Highlights
The impact of COVID-19 lockdown on HIV care in 65 South African primary care clinics: an interrupted time series analysis
(The Lancet HIV, March 2021)
Main Message: In South Africa, among the most concerning disruptions due to COVID-19 control measures, is the ability to avail of health care unrelated to COVID-19— particularly in light of the hard-won progress toward mitigating the burden of infectious diseases like HIV. A study of 65 primary health clinics in South Africa showed that while antiretroviral treatment (ART) was generally maintained, there was a significant decrease in HIV testing and ART initiation during the time of the lockdown between April and July 2020. Given that disruptions to ART provision is a primary driver of morbidity and mortality among people with HIV, the success of continuing ART treatment for those already linked to care is encouraging. Even so, the study showed that people infected with HIV but not linked to care were among the most severely affected by the lockdowns, particularly since they are likely at elevated risk of severe COVID-19 infection. COVID-19-related disruptions to ART supply chains and future COVID-19 outbreaks still pose significant threats to progress made by HIV programs, particularly in countries with more precarious health infrastructure.
- This study analyzed data from 65 public sector clinics in KwaZulu-Natal, South Africa. This province had the third most reported COVID-19 cases in South Africa, alongside an HIV prevalence of 27% among adults aged 15-49.
- Researchers included data from people testing for HIV, initiating ART and collecting ART prescription refills at participating clinics between January 2018 and July 31, 2020. This interrupted time series analysis compared the mean and median number of monthly tests, ART initiations and prescription refills from January 2018–March 2020 (27 months prior to the COVID-19 lockdown) and April–July 2020 (4 months during the lockdown).
- The study showed a 47.6% decrease in monthly HIV testing and a 46.2% decrease in monthly ART initiation during the 2020 COVID-19 lockdown compared to pre-lockdown rates, accounting for seasonality and pre-lockdown trends.
- Data were not available to calculate weekly testing, initiation and prescription refill trends, which could be more informative given the dynamic nature of lockdown measures. The researchers were also not able to analyze HIV viral load outcomes which could help point more definitively to the effects of delaying linkage to HIV care and treatment.
Suggested citation: Cash-Goldwasser S, Jones SA, Bochner A, Cobb L and Frieden TR. In-Depth COVID-19 Science Review March 18 – April 16, 2021. Resolve to Save Lives. 2021 April 15. Available from https://preventepidemics.org/covid19/science/review/
