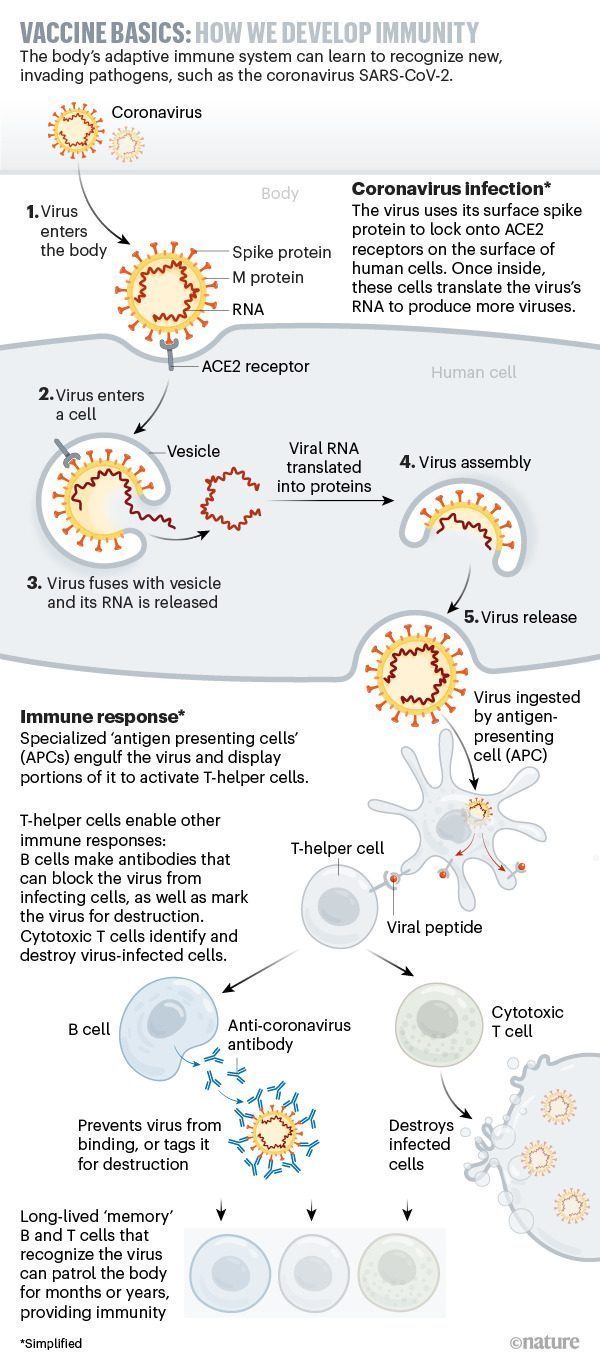
In-depth
Update on COVID-19 vaccine development
There is tremendous global interest in a vaccine to prevent infection or severe disease with SARS-CoV-2, the virus that causes COVID-19, as this could be the most effective tool to curb the COVID-19 pandemic. Vaccination exposes the immune system, a highly complex infection-fighting array of organs, cells and proteins, to the pathogen of interest, thus generating immunity. Immunity can also be generated through natural infection, although for SARS-CoV-2, it is not yet clear how much or for how long previous infection protects against reinfection. Even under the assumption that previous infection provides some protection, the proportion of the population that must be immune in order to stop the spread of infection (a concept referred to as “herd immunity”) can be difficult to achieve through natural infection alone. Multiple studies (including a recently published seroprevalence study conducted in the U.S., reviewed below) have shown that population seroprevalence levels are still relatively low. In addition, natural infection is associated with significant risks. A good vaccine would teach the immune system to fight SARS-CoV-2 without causing infection and without resulting in other types of harm. Ideally, a COVID-19 vaccine would be effective after one dose without need for additional (booster) doses, as fewer doses reduce the financial and human resource requirements of any vaccination program. In addition, an ideal vaccine would protect target vulnerable populations such as older adults and those with comorbidities that confer risk of severe COVID-19, while also being safe in those populations, including for immunocompromised people. An ideal COVID-19 vaccine would confer protection for as much time as possible before immunity levels wane, as they can over time. In addition, an ideal vaccine would reduce onward transmission of the virus to contacts; some vaccines may protect against severe clinical disease but still allow vaccinated people to become infected and thus, to potentially spread the infection. It is unlikely that the first COVID-19 vaccine will be ideal in all of these ways.
For a vaccine to gain approval and licensure—whether from the Food and Drug Administration (FDA) in the United States, the European Medicines Agency (EMA) in Europe, or other regulatory bodies in other countries or regions—at least three phases of clinical testing are generally completed. Before clinical testing, a vaccine is typically tested in animals. Those animals are monitored for harm the vaccine may cause, and their immune responses to the vaccine are measured; in addition, those animals may be exposed to the pathogen to assess whether the vaccine has generated protective immunity. Over 140 candidate COVID-19 vaccines are currently undergoing pre-clinical testing in animals, and 25 vaccines have progressed to clinical testing. The first required phase of clinical testing (Phase I) typically includes fewer than 100 healthy volunteers and is designed to assess whether the vaccine stimulates the immune system, as well as to monitor safety and determine a dosage that both stimulates the immune system and is safe. The second required phase of clinical testing (Phase II) typically lasts at least several months and includes several hundred volunteers in order to assess whether the chosen vaccine dose stimulates the immune system and is safe in more people, including people who may differ by age, race, ethnicity, sex, underlying health status and other characteristics. The final required phase of clinical testing (Phase III) typically includes thousands of volunteers. In this phase, there is safety monitoring over an even longer period of time and an assessment of the ability of the vaccine to prevent natural infection. These three phases typically take years to complete, and the majority of vaccine candidates do not successfully progress through the process. To shorten the timeline of vaccine approval during an epidemic or pandemic, trial phases may be combined or researchers may use a “pandemic paradigm” in which several steps that would typically be executed in succession are instead executed in parallel.

After the conclusion of Phase III trials, regulators review trial data and determine whether to approve the vaccine for use outside a clinical trial (in some cases pre-licensure use may be approved; in the U.S., the FDA can allow “compassionate use” or grant emergency use authorization). In the United States, the Advisory Committee on Immunization Practices (ACIP) then recommends vaccine prioritization, dosage, and recommendations for different groups, based on a combination of information about the vaccine and information about the epidemiology of naturally occurring illness. After a vaccine is approved and made available to the public, a Phase IV trial may be conducted during which large-scale, long-term safety and efficacy data are collected. Even if a Phase IV trial is not conducted, passive surveillance systems are used to monitor adverse events, including rare, longer-term adverse events that could not be observed in earlier trials. Safety in each phase is assessed by examining for “adverse reactions” or undesired harmful effects resulting from the vaccine. Adverse reactions may be local (i.e., pain, redness or swelling at the injection site) or systemic (e.g., fever or muscle aches) and may be mild (e.g., mild fatigue) or severe (e.g., anaphylaxis). Generally, vaccine efficacy can be assessed two ways. This first is to perform blood tests in a laboratory to detect elements of immunity. For COVID-19 and other infectious diseases, these tests typically involve measuring antibodies such as neutralizing antibodies (proteins produced by immune cells which stick to and stop viruses from infecting host cells) and measuring immune cells such as T-cells (a type of immune cell that destroys infected cells and helps provide long-lasting immunologic memory). The second way is to determine whether those who are vaccinated are protected from natural infection. It is unclear to what degree these two types of measures for protection against COVID-19 are interchangeable. Related to this, there is evidence that may suggest that antibodies to SARS-CoV-2 wane significantly during the months following infection, but this may not mean that protection against infection is lost.
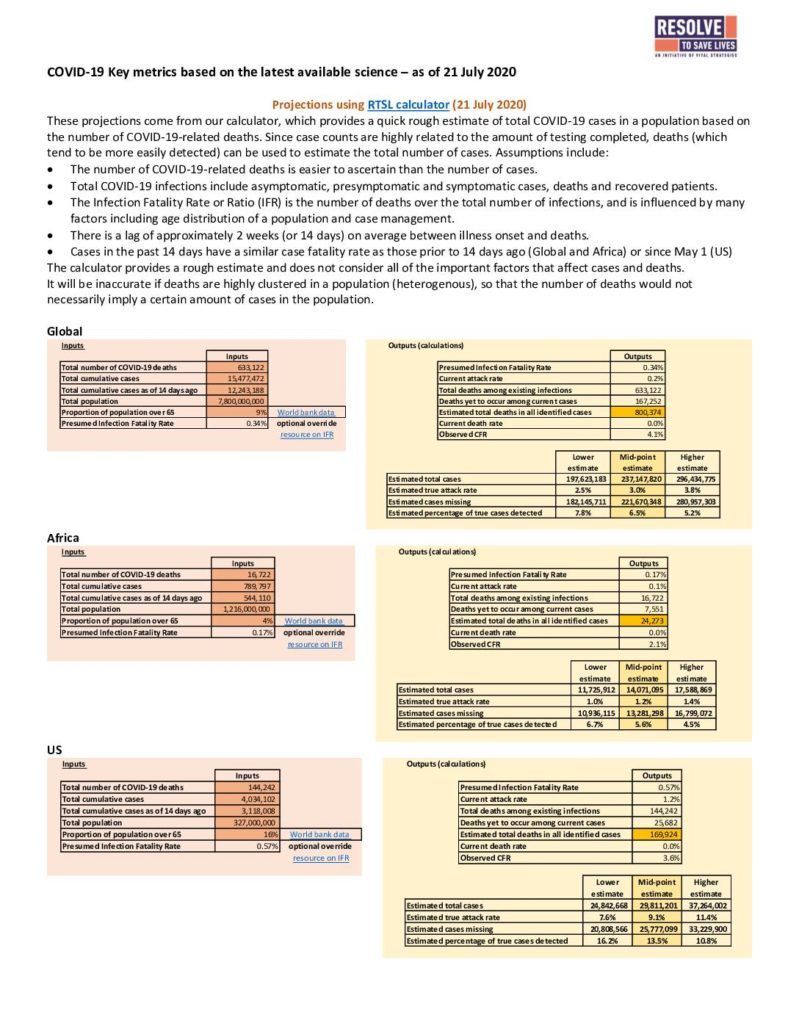
The figure below depicts how the human immune system can learn to recognize new pathogens such as the coronavirus that causes COVID-19.
Source: The race for coronavirus vaccines: a graphical guide
Scientists are using a number of vaccine platforms to develop COVID-19 vaccines. One platform, used by many currently licensed vaccines (including measles, smallpox, some polio and some influenza vaccines), contains inactivated or weakened (“live attenuated”) whole virus. There are platforms that use newer molecular technologies, including genetic vaccines that deliver pieces of viral DNA or RNA into host cells. These genetic fragments, which produce proteins to which the immune system may respond, are not capable of causing an infection. Although there are no genetic vaccines currently licensed for use in humans, several promising COVID-19 vaccine candidates poised to enter Phase III trials (discussed below) are based on this platform. There are also vaccines that use the cell-entry machinery of a virus that cannot cause disease as a vector to deliver proteins of the target virus into host cells so that immunity to the target virus is generated. The viral vectors used in such vaccines can be non-replicating or replicating. A non-replicating vector has been modified so it cannot multiply, so each individual vector virus particle (virion) delivers the target virus proteins into one cell. A replicating vector can generate multiple vector virions carrying the target proteins, so multiple cells can receive the target protein and more immunity can be generated, potentially reducing the need for additional vaccine doses. The Ebola vaccine approved by the FDA in 2019 uses a replicating viral vector.
Results of Phase I and II clinical trials for two non-replicating viral vector COVID-19 vaccines have recently been published in The Lancet. Those results support progression to Phase III trials. One trial, a Phase II trial conducted at a single center in Wuhan, China, tested a COVID-19 vaccine candidate that uses human adenovirus type 5 (Ad5), which may cause mild respiratory in humans in its natural state, as a vector to deliver a SARS-CoV-2 protein. In this study, 508 healthy adults were randomly allocated to receive either the Ad5-vectored COVID-19 vaccine (in one of two doses that were chosen based on the results of the Phase I trial), or placebo. There was no age cap; 13% of participants were age 55 or older. After 28 days of follow-up, approximately 85% of COVID-19 vaccine recipients generated neutralizing antibodies and over 90% had T-cell responses to SARS-CoV-2. Adverse events were more common in those who received the COVID-19 vaccine than among placebo recipients, but no serious adverse events occurred. Notably, immune responses were not as strong in older participants, which may indicate a need for a booster dose in some subpopulations. Immune responses were also not as strong in participants who had evidence of immunity to Ad5; this is potentially an issue when a viral vector to which many people have already been exposed is used. The other trial, a Phase I/II trial conducted at five sites in the United Kingdom, was on a COVID-19 vaccine candidate that uses a chimpanzee adenovirus vector developed at the University of Oxford (ChAdOx1) to express a SARS-CoV-2 protein. In this study, 1,077 healthy adults under age 55 were randomly allocated to receive either the ChAdOx1 nCoV19 vaccine or a vaccine against meningitis. Adverse events were more common among COVID-19 vaccine recipients but no serious adverse events occurred. More than 90% of COVID-19 vaccine recipients generated neutralizing antibodies to SARS-CoV-2 and responses were sustained for up to 56 days, which was the final point of data collection before results were published. T-cell responses were induced in all participants. For both vaccines, strong immune responses in all or the vast majority of participants and the absence of severe adverse events support progression into Phase III trials. Though this is cause for some optimism, inferences about vaccine efficacy and safety should be made cautiously for several reasons, including that: the number of participants evaluated was small; cohorts were not racially diverse; there has not yet been an assessment of whether vaccination prevents infection in humans; and there has not been significant longitudinal immunologic or safety follow-up. The study in China plans to follow participants for six months and the study in the United Kingdom plans to follow participants for one year.
Phase III trials for the ChAdOx1 vaccine are underway in Brazil, South Africa and the United Kingdom. Generally, Phase III trials are large and inclusive enough to assess many aspects of vaccine safety and real-world efficacy in subgroups of interest. In addition, Phase III trials can answer questions about whether a single dose is sufficient in older adults and other subgroups, about how much measured immune responses correlate with protection against infection, and about the longevity of immunologic protection. It is important that vaccine safety and efficacy are assessed in diverse populations across a range of settings. Vaccine efficacy may differ between high- and low-income settings. Specifically including low-income settings in international clinical trials is often avoided or overlooked, sometimes for scientific reasons but sometimes for financial or ethical ones. It is also important that the incidence of disease is high enough in the study area that robust data to support firm conclusions can be gathered in a reasonable amount of time. Another COVID-19 vaccine candidate produced in China, an inactivated vaccine, is undergoing a Phase III trial in Brazil after reportedly promising Phase I/II results. Promising results of a Phase I trial, conducted in the U.S. on a COVID-19 genetic vaccine candidate known as mRNA-1273, were recently published and Phase III trials of this vaccine and still another genetic COVID-19 vaccine candidate are expected to begin recruitment in the U.S. soon.
There are multiple complex aspects of vaccine development, testing, production, allocation, and implementation beyond the scientific ones. One is the financial investment. This is especially true for vaccine development during a pandemic, as manufacturing and distribution systems must be put in place while vaccine candidates are still being developed and tested in order to avoid delays once a vaccine is approved for use. This can entail significant financial risk. Enormous sums of private and public money and significant resources have been allocated to support COVID-19 vaccine research and development. For example, Operation Warp Speed, a public-private partnership initiated by the U.S. government that strategizes to accelerate the development of COVID-19 countermeasures, has provided massive financial support toward the stated aim of delivering 300 million doses of a safe, effective COVID-19 vaccine by January 2021. However, there is concern that such a single-nation approach to vaccine development, without global coordination and without investment by multiple countries, will lead to serious and damaging inequities in COVID-19 vaccine access, particularly for low-income countries. In addition, there are questions of who will be prioritized to receive any new COVID-19 vaccine within a single country, as supplies of any vaccine will be initially limited. Another concern is that mistrust of a COVID-19 vaccine could imperil the prospect of achieving sufficient coverage for herd immunity even if a safe and effective vaccine does become available. This issue was discussed during a U.S. congressional hearing in late June. Experts argue that the best response to such concerns is a transparent and rigorous approach to vaccine development and regulation, including publicly disseminated data demonstrating strong evidence of effectiveness, strong evidence of safety, caution around pre-licensure use, and comprehensive safety monitoring systems. It is important that this concern is addressed proactively, rather than waiting for vaccine hesitancy to become a problem once a safe and effective COVID-19 vaccine becomes available.
FAQs
What is antigen testing?
An antigen test is a type of diagnostic test that detects the presence of an agent or substance such as a virus, bacteria or chemical by identifying specific proteins or molecules that usually mark the outside of the agent. These proteins or molecules are part of the agent’s antigen; a substance that is capable of producing an immune response in the body. Antigen diagnostic tests differ from molecular diagnostic tests such as reverse-transcriptase polymerase chain reaction (RT-PCR) in that they do not identify genetic material and they can be processed at the point of care, outside of a traditional laboratory. In addition to being faster than molecular diagnostic tests, antigen tests can also be less costly. However, they sometimes lack the accuracy of molecular tests and can give false negative results, requiring additional followup.
Many people are likely more familiar with antigen testing than they realize, and may have even previously been tested for an infection using this kind of rapid, point-of-care test. Antigen tests including the rapid flu and rapid strep tests are commonly used in medical offices, urgent care facilities and emergency departments to provide quick and reliable diagnostic results. Results are typically delivered in less than an hour and sometimes in less than 15 minutes. Adding such capacity to COVID-19 testing can improve the time it takes to get results, decrease the burden placed on strained laboratories, and scale up diagnostic testing, which is one of the pillars of COVID-19 response. It can potentially also improve public health capacity to isolate cases quickly and accelerate contact tracing efforts. To date, the U.S. Food and Drug Administration (FDA) has approved two antigen tests for COVID-19: The Quidel Corporation received the first emergency use authorization in May 2020, and BD (Becton Dickinson) received the second emergency use authorization in July. To keep the public informed about the different types of tests currently available for COVID-19, the FDA and other agencies such as the U.S. Centers for Disease Control and Prevention (CDC) have updated their information on diagnostic tests to include antigen tests. In general guidance for COVID-19 testing, the FDA states that positive results from antigen tests ”are usually highly accurate but negative results may need to be confirmed” with a molecular test in some situations, such as when someone has symptoms of COVID-19.
| Molecular Test | Antigen Test | Antibody Test | |
|---|---|---|---|
| Also known as… | Diagnostic test, viral test, molecular test, nucleic acid amplification test (NAAT), RT-PCR test, LAMP test | Rapid diagnostic test (Some molecular tests are also rapid tests.) | Serological test, serology, blood test, serology test |
| How the sample is taken… | Nasal or throat swab (most tests) Saliva (a few tests) | Nasal or throat swab | Finger stick or blood draw |
| How long it takes to get results… | Same day (some locations) or up to a week | One hour or less | Same day (many locations) or 1-3 days |
| Is another test needed… | This test is typically highly accurate and usually does not need to be repeated. | Positive results are usually highly accurate but negative results may need to be confirmed with a molecular test. | Sometimes a second antibody test is needed for accurate results. |
| What it shows… | Diagnoses active coronavirus infection | Diagnoses active coronavirus infection | Shows if you’ve been infected by coronavirus in the past |
| What it can’t do… | Show if you ever had COVID-19 or were infected with the coronavirus in the past | Definitively rule out active coronavirus infection. Antigen tests are more likely to miss an active coronavirus infection compared to molecular tests. Your health care provider may order a molecular test if your antigen test shows a negative result but you have symptoms of COVID-19. | Diagnose active coronavirus infection at the time of the test or show that you do not have COVID-19 |
Source: Coronavirus Testing Basics (FDA)
What is pooled testing?
As COVID-19 cases surge and the number of people seeking testing increases in the U.S., laboratory capacity has not been able to meet demand for diagnostic testing. In some parts of the country, especially the South, Southwest and West, test results can take as long as two weeks. This delay results in a missed opportunity to isolate positive cases and their contacts in a timely manner and prevent ongoing transmission. It can also further disrupt the lives of people who are not infected and would have otherwise been able to continue their day-to-day activities with appropriate “3W” measures such as wearing a mask, washing their hands and watching their distance. Pooled testing (sometimes called batch testing) may be one approach that communities can use to address strained testing capacity.
Typically, when someone submits a swab for a diagnostic COVID-19 test, the sample is processed as an individual specimen through a machine that tests for the presence of genetic material from the virus. These machines can become a bottleneck for turnaround time due to limitations on how many tests they can perform at a time. Pool testing refers to a process where multiple samples from different people are pooled (combined) and run as a single test. By pooling tests, a laboratory that would normally be able to run 100 tests in a day can scale up to 500 tests a day. Pool testing is already used for other diseases and in specific settings such as the screening of blood donations. When a pooled specimen is processed and the results are negative, this means that all of the samples included in the pool are negative and can be reported immediately in a fraction of the time and cost that it would have taken to run each specimen individually. If the results of a pooled specimen are positive, it means that at least one of the samples included in the pool was positive, and those samples will each have to be retested individually. For this reason, pooled testing works best when the rate of positive tests in a population is low; it may not be appropriate or efficient in all settings. It would not be effective, for example, in a setting where transmission is high and many symptomatic people are being tested. It could be useful, in a setting where overall community transmission is low, or when testing for surveillance or screening (e.g. in a factory or for health care workers) is adding to the overall testing volume of a community, and a high proportion of positive tests is not expected. Federal officials in the U.S. have included pooled testing as one strategy to address current testing capacity shortages. Pooled testing may also play a larger role in screening efforts in the future, including when testing prioritization is expanded to include testing for asymptomatic people who do not have a known COVID-19 contact. Pool testing may not be the answer for communities in the U.S. currently experiencing high test positivity and a surge in cases, as many of the samples would need to be retested.
Weekly Research Highlights
Seroprevalence of Antibodies to SARS-CoV-2 in 10 Sites in the United States, March 23-May 12, 2020
(JAMA, July 21)
-
Researchers collected and tested 16,025 anonymized lab specimens that were obtained from patients for reasons unrelated to COVID-19 between March 23 and May 12 in 10 sites across the United States. People over 65 years old accounted for the highest proportion of specimens (37%), while people under 18 years old accounted for the lowest proportion (8%). Women accounted for 55% of the specimens. Due to variation in demographic characteristics, final results were presented in unadjusted as well as age-sex standardized estimates.
-
Age-sex adjusted overall seroprevalence for each of the 10 sites was as follows: Western Washington State—1.1%; New York City metro area—6.9%; Louisiana— 5.8%; South Florida—1.9%; Philadelphia metro area—3.2%; Missouri—2.7%; Utah—2.2%; San Franscisco Bay area—1.0%; Connecticut—4.9%; Minneapolis-St. Paul-St. Cloud area—2.4%. These estimates for infections range from six to 24 times higher than the number of infections recorded from reported cases alone, but closer to 10 times higher in seven of the 10 jurisdictions.
-
The study’s limitations include that the samples used were obtained from people seeking health care and may not be representative of the general population in each location. In addition, no information about whether people had recent illness or symptoms was available. The antibody test used has limitations in sensitivity and specificity, resulting in an unknown proportion of false positive and false negative test results, which may affect the overall seroprevalence proportions. These results cannot speak to any type of protective immunity following SARS-CoV-2 infection. The seroprevalence proportions reported in this study are based on the timing of the testing done at each site, and do not represent the current expected value.
-
Though not reported in this study, a second round of testing was performed in some of the sites as part of ongoing commercial laboratory serosurveillance. In the New York City metro area, the overall seroprevalence rose to 23.3% in late April and early May, compared to 6.9% in late March, as was reported in this study. Repeat testing from seven other sites showed only slightly higher or similar results to the first round of testing.
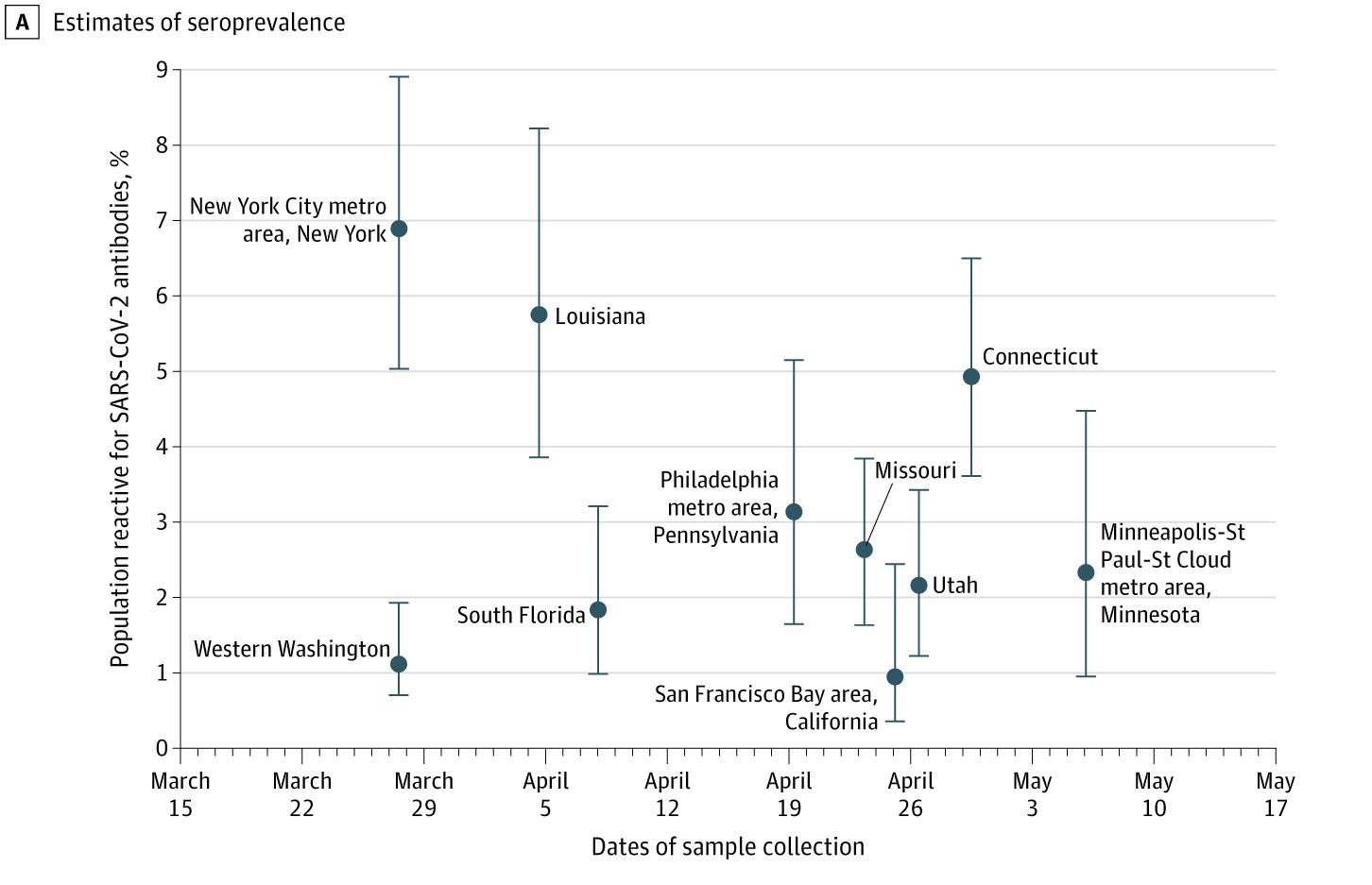
CDC MMWR A, Estimates are shown with 95% confidence intervals for 10 geographic sites from which residual clinical specimens were collected. Seroprevalence estimate is shown at the midpoint of the specimen collection date range. March 27 to May 6.
Impact of delays on effectiveness of contact tracing strategies for COVID-19: a modelling study
(Lancet Public Health, July 16)
-
Researchers adapted a stochastic model to simulate the impact of adding contact tracing to public health and social measures on COVID-19 transmission. They compared how varying levels of coverage and delays in testing (interval between symptom onset and diagnosis) and tracing (interval between confirming an index case and tracing all of their contacts) could affect transmission. With 80% to 100% coverage and minimal delays at each step, adding contact tracing could reduce the effective reproductive number (Rt) by 17% or more and effectively suppress the outbreak (keeping Rt<1.0).
-
Delays at either step could reduce the impact of contact tracing, but testing delays were the most important factor. The proportion of transmission events prevented ranged from 79.9% when testing delay was less than a day, to 41.8% with a three-day testing delay and 4.9% with a seven-day delay, given optimal coverage and minimal tracing delay.
-
Researchers assumed that mobile apps could improve tracing coverage and reduce tracing delays compared to conventional contact tracing. As a result they estimated that conventional contact tracing would only keep Rt<1.0 when testing delay was less than one day, whereas mobile app technology could suppress transmission even when testing delays of one or two days were present.
-
Contact tracing can contribute to suppressing COVID-19 transmission even as some public health and social measures are lifted, provided the contact tracing program optimizes coverage and limits delays at each step, particularly testing. Some of the assumptions underlying this model would be affected by changes in testing or improved knowledge about the role of asymptomatic infections in transmission of COVID-19. Although some places such as Taiwan and South Korea have used mobile apps to improve the coverage and timeliness of contact tracing as described in this simulation, it is not possible to attribute their success controlling COVID-19 to these tools.
Notes from the field: Effects of the COVID-19 Response on Tuberculosis Prevention and Control efforts — United States, March–April 2020
(MMWR, July 24)
-
Researchers at the U.S. CDC communicated with 50 of 61 jurisdictions that receive funding for TB control and prevention activities across the country about the impact of resource diversion on TB elimination activities. They asked about changes to staffing capacity for various efforts as well as each program’s ability to continue essential activities.
-
Many staff were transferred from their roles in TB control and prevention to participate in the U.S. COVID-19 response, and most jurisdictions reported partial or high impact on their TB capacity and activities as a result. All aspects of TB control and prevention were affected, including diagnosis and treatment, surveillance, contact tracing, education and training, and other administrative and field activities. Though direct effect on outcomes was not measured in this study, the diversion of resources from TB control to COVID-19 may affect the number of cases and those who are able to complete treatment.
-
TB control is one of the country’s essential public health services that will likely need to address a backlog of activity due to diversion of resources to COVID-19.
Suggested citation: Cash-Goldwasser S, Kardooni S, Cobb L, Bochner A, Bradford E and Shahpar C. In-Depth COVID-19 Science Review July 18 – 24, 2020. Resolve to Save Lives. 2020 July 28. Available from https://preventepidemics.org/covid19/science/review/